
The Evolution of Improved Fitness
by
random mutation plus selection
Copyright © 1999 by
Edward E. Max, M.D.,
Ph.D.
[Last Update: July 12,
1999]
![]()
The Evolution of Improved Fitness
by
random mutation plus selection
Copyright © 1999 by
Edward E. Max, M.D.,
Ph.D.
[Last Update: July 12,
1999]
![]()
|
Correspondence with a Critic In June of 2000, a reader of this Talk.Origins essay suggested that I solicit a response from Dr. Lee M. Spetner, whose book "Not by Chance" attacks the theory of evolution using arguments from information theory, in an effort to support a poorly articulated "creation" alternative. I emailed Spetner, and an extensive (and interesting, in my view) correspondence ensued, which we agreed to link to the present article. The unedited correspondence is difficult to follow. There were no length limitations placed on either of us, and many points made by each of us were addressed by the other in comments spread over several successive Emails. To make the correspondence readable I have assembled together the various arguments relevant to a particular issue in the debate. My edited version is linked here. This version contains most of the arguments central to the correspondence, but due to time constraints, some have been omitted. This edited versiion has not been approved by Spetner, and he will likely desire changes and additions. The correspondence will be updated periodically as time permits. |
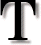 he theory of evolution includes a number of ideas that
some people find difficult to accept intuitively. One of the most difficult
seems to be the notion that the intricate and interdependent structures we
observe in modern plants and animals arose through random genetic mutations
selected over time. For some people it is much easier to believe that the
beautiful and functional features of the human eye, for example, were designed
by an intelligent creator than to imagine how they could have been generated
through random events. Creationists capitalize on this conceptual difficulty,
presenting several arguments that appear to demonstrate that random mechanisms
could never lead to even a single functioning protein, let alone an eye. These
arguments can be refuted by theoretical counter-arguments; yet many people have
difficulty accepting these counter-arguments at an intuitive level. What might
be persuasive would be a clear example in living organisms that shows how random
mutation plus selection can lead to improved "fitness." Some time ago I realized
that just such an example was provided by experiments related to my own
laboratory research, which concerns the genes encoding the immune system
proteins known as antibodies. Because antibody genes are not well known to the
general public, I decided to write this article in the hope that it might be
useful for readers perplexed by the creationist arguments.
he theory of evolution includes a number of ideas that
some people find difficult to accept intuitively. One of the most difficult
seems to be the notion that the intricate and interdependent structures we
observe in modern plants and animals arose through random genetic mutations
selected over time. For some people it is much easier to believe that the
beautiful and functional features of the human eye, for example, were designed
by an intelligent creator than to imagine how they could have been generated
through random events. Creationists capitalize on this conceptual difficulty,
presenting several arguments that appear to demonstrate that random mechanisms
could never lead to even a single functioning protein, let alone an eye. These
arguments can be refuted by theoretical counter-arguments; yet many people have
difficulty accepting these counter-arguments at an intuitive level. What might
be persuasive would be a clear example in living organisms that shows how random
mutation plus selection can lead to improved "fitness." Some time ago I realized
that just such an example was provided by experiments related to my own
laboratory research, which concerns the genes encoding the immune system
proteins known as antibodies. Because antibody genes are not well known to the
general public, I decided to write this article in the hope that it might be
useful for readers perplexed by the creationist arguments.
Before discussing antibody genes, it is worthwhile to consider what the creationists say about randomness versus design. Let's focus on three favorite arguments of the creationists which seem persuasive because they contain some true elements and some valid logic.
First, creationists proclaim that mutations are harmful. According to the creationists, if you take a well-running complex biological machine and subject it to random alterations, you could scarcely expect to have made any improvements and almost certainly will have harmed the organism. Creationists point out that all the classic mutations seen in humans are detrimental, causing such genetic diseases as sickle cell anemia, muscular dystrophy, cystic fibrosis, and cancer syndromes. Not a single clear example of a beneficial human mutation has been described. To explain adaptive changes in populations -- such as the famous darkening of the peppered moth population (which occurred when the soot-darkened trees resulting from heavy industry made light-colored moths easier targets for hungry birds) -- the creationists argue that the variant genes for dark and light color were present in the original population, designed by a creator to allow moths to live in varying environments; in this view the shift towards darker coloration in the moth population resulted from shifts in the frequencies of existing designed genes without requiring new random mutations. According to creationists, the essential relationship between natural selection and mutations is that selection acts conservatively to weed out individuals with mutations and to prevent the spread of mutations into a population.
Several formal arguments have been put forward to the effect that random mutations cannot increase the information content of a system. Since the information content of the human genome is vastly larger than that of bacteria, if mutation cannot play a role in this increase then the foundation of evolution by mutation and natural selection would seem to be in question.
A final argument put forward by creationists against the role of random mutation in biological origins is a statistical calculation supposedly proving the impossibility of an evolutionary origin of proteins. A protein is large biological molecule constructed of smaller subunits, known as amino acids, which become linked together in a linear chain and then folded into a precise three-dimensional structure. There are twenty different kinds of amino acids in proteins, and the specific sequence of these amino acids determines the final shape and properties of a given protein. A typical protein is composed of a hundred or more amino acids in a strictly defined order. The creationists ask: what is the probability that the correct sequence of amino acids of a specific protein -- for example, the 141 amino acids of the human oxygen-carrying protein called globin -- could have been selected by chance? The total number of possible 141-long amino acid sequences is 20141, a number so incomprehensibly huge that the statistical probability of the actual globin sequence ever appearing from a random assortment of amino acids is negligibly small. In a picturesque simile, the creationists are fond of comparing the probability of correctly assembling a protein sequence by this random selection model to the likelihood that a tornado blowing through a junkyard could assemble a 747 airliner.
While it is true that most mutations are either harmful, as suggested by the creationists, or neutral, the creationists gloss over a crucial fact: beneficial mutations do occur, though they are very rare. Can a beneficial mutation that occurs once in million individuals ever really contribute to evolution? Yes it can, since a rare beneficial mutation can confer a survival or reproductive advantage to the individuals that carry it, thereby leading -- over several generations -- to the spread of this mutation throughout a population. Beneficial mutations occurring in several different individuals in several different genes can simultaneously spread through a population, and can be followed by successive rounds of additional mutation and selection.
Does the fact that we know many human detrimental mutations but essentially no clear beneficial ones mean that there are have been no beneficial mutations in human history? Not at all, since there is a clear bias in what medical scientists have studied. The human mutations we know most about are detrimental because medical scientists preferentially study illnesses that cause significant morbidity and mortality. Consider the theoretical possibility that a beneficial mutation has occurred in a particular human gene; even if this mutation were identified by a comparison of the mutated gene in a child versus the unmutated version of the same gene in both parents, there is no way that this mutation could ever be recognized as beneficial. If the mutation increased intelligence, strength, longevity or specific disease resistance, this would never be apparent without long-term breeding experiments that could obviously never be done on humans. Therefore, since such beneficial mutations in humans could never be recognized in humans, our ignorance of examples cannot be taken as evidence that they don't exist. However, the experiments necessary to demonstrate a beneficial mutation can be done with laboratory organisms that multiply rapidly, and indeed such experiments have shown that rare beneficial mutations can occur. For instance, from a single bacterium one can grow a population in the presence of an antibiotic, and demonstrate that organisms surviving this culture have mutations in genes that confer antibiotic resistance. In this case (in contrast to the situation with the peppered moth populations described above) origin of the population from a single bacterium allows comparisons of the mutated genes with the corresponding genes from the original bacterium, verifying that the variant sequences were not present before the culture with antibiotics and therefore arose as de novo beneficial mutations.
While a detailed mathematical consideration of information theory is beyond the scope of this article, none of the creationist arguments based on information theory that I am aware of adequately address the obvious increase in information that can occur when a gene duplicates and the two copies undergo independent mutations leading to two genes with somewhat different functions. Gene duplication, mutation and selection are all known to occur due to natural biochemical processes in a variety of organisms studied in the laboratory. Many gene families are known with members that encode proteins having related structure and related but distinct function. Each family can be explained by multiple gene duplications followed by random mutation and differentiation of the functions of the individual gene copies. Clearly the expansion from a single primordial gene to a large family of genes with distinct functions represents an increase in genetic information.
An example that I have already mentioned in another posting on Talk.Origins is the hemoglobin/myoglobin family. The gene for a primordial oxygen-carrying protein is thought to have duplicated leading to separate genes encoding myoglobin (the oxygen-carrying protein of muscle) and hemoglobin (the oxygen-carrying protein of red blood cells). Then the hemoglobin gene duplicated, and the copies differentiated into the forms known as alpha and beta. Later, both the alpha and beta hemoglobin genes duplicated several times producing a cluster of hemoglobin-alpha-related sequences and a cluster of hemoglobin-beta-related sequences. The clusters include functional genes that are slightly different, that are expressed at different times during the development of the embryo to the adult, and that encode proteins specifically adapted to those developmental periods. Other examples of gene families that appear to have developed by such duplication and differentiation include the immunoglobulin superfamily (comprising a large variety of cell surface proteins), the family of seven-membrane-spanning domain proteins (including receptors for light, odors, chemokines and neurotransmitters), the G-protein family (some members of which transduce the signals of the seven-membrane-spanning domain family proteins), the serine protease family (digestive and blood coagulation proteins) and the homeobox family (proteins critical in development). A large part of the increase in information in our genomes compared with those of "lower" organisms apparently results from such gene duplication followed by independent evolution and differentiation of duplicated copies into multiple genes with distinct function. If an information theory analysis claims that random mutation cannot lead to an increase in information but the analysis ignores gene duplication and differentiation through independent mutations, such an analysis is irrelevant as a model for gene evolution, regardless of its mathematical sophistication.
|
While my purpose in this posting is not to defend Dawkins but to describe a different, biological model of mutation and selection, so many readers have written to offer the same two additional concerns about Dawkins's simulation that I feel obliged to discuss them. One concern is that Dawkins's target sequence was specified in advance by an intelligent designer, and the other is that Dawkins's simulation does not correspond well to biological evolution. I will try to present my own views of these two concerns in turn. The fact that the target sequence in the "weasel" model was chosen in advance is an unavoidable consequence of Dawkins's goal of contrasting the single-step selection model with the model of multiple sequential rounds of mutation and selection. The creationist single-step model also starts with a specific protein sequence and then tries to calculate the odds of ever achieving this target sequence by a single selection step from random sequences; thus, a target sequence is specified in advance for either model. (Incidentally, whether this target sequence originally derived from an intelligent design or an evolutionary process is irrelevant to the question of whether it can be created anew by a procedure involving randomness and selection. In particular, Dawkins's choice of a 28-letter line from Shakespeare rather than a 28-residue amino acid sequence from a known protein is irrelevant in judging the relative efficacies of the two procedures in reproducing the target sequence.) The result of Dawkins's exercise is to show that the single-step procedure is essentially incapable of arriving at the target sequence within a reasonable timeframe, while the sequential-step procedure can readily achieve the same pre-specified target sequence. This difference is all that Dawkins was trying to show. Thus the complaint that "specification of the target sequence in advance weakens Dawkins's result" is not valid, in that this same feature (specifying a target sequence in advance) is applied equally to both models, as it must be to compare their efficacy. How well does Dawkins's model simulate evolution? In thinking about this question, one needs to specify whether the simulation is modeling abiogenesis (the origin of life from non-life) or evolution from pre-existing life forms. Almost nothing is known about abiogenesis, and the mechanisms involved are highly speculative; in contrast, a large body of evidence supports evolution by common descent, with natural selection playing a significant role. Dawkins's model doesn't seem to correspond exactly to either abiogenesis or evolution. The model supposes that a mechanism exists for replicating existing copies of the sequence to produce a "progeny" generation (how such a replication mechanism may have arisen is not addressed by the model). A replication mechanism in the model suggests that the simulation is a model for a process in evolution after life -- including reproduction -- has already arisen. But once life has arisen, evolution of a protein from random amino acid sequences is not thought to be the usual mechanism by which new proteins arise in evolution; instead, new proteins are believed to arise by modification of pre-existing protein sequences, usually after gene duplication. However, some proteins with essentially random amino acid sequences are generated by frame-shift mutations (causing a coding sequence to be read in the wrong triplet reading frame) or by splicing mutations (that convert non-coding intron sequence into coding sequence). These processes might have occurred more frequently an early stage of "proto-life," at a time when a replication mechanism had arisen, but was less accurate than replication is today. A critical component of Dawkins's model is that one of these random protein sequences could have some tiny activity that was useful in some way, probably by interacting with other materials in its environment, allowing the protein to provide a selective advantage that would tend to increase its frequency in later generations by natural selection. Then each successive mutation could have provided a slight increase in efficiency of that function that would allow the mutated protein to replace earlier versions of the protein through further selection. In this model, each step must provide an increase in efficiency, in contrast to the creationist idea that there can be no selectable function until the final sequence is reached. Thus, although Dawkins's simulation does not match closely to the usual model of evolution of a protein from a pre-existing sequence, it does correspond to one possible scenario in the context of evolutionary theory. In contrast, the single-step selection model does not correspond to anything in evolutionary theory. This model is exploited by creationists because they can claim to naive audiences that it is implied by the evolutionist terms "random" and "selection," after which they can demolish this straw-man by demonstrating the impossibility of its achieving a target protein sequence. They avoid mentioning that an alternative model involving "random" and "selection" -- the sequential multi-step model, which more accurately reflects evolutionary theory -- can easily achieve a target sequence. |
What about the argument concerning the statistical improbability of obtaining a specific 141 amino acid sequence by looking for the correct sequence among randomly generated sequences? Certainly this mechanism could not explain the origin of protein sequences, but the creationist suggestion that this mechanism is part of evolutionary theory is false; it is a "straw-man" -- a false creationist caricature of evolution -- used repeatedly by creationists to mislead naive audiences into thinking that evolution is illogical. It is false because it demands a specific sequence in a SINGLE selection step from a pool of random sequences, whereas the real evolutionary model for the origin of protein sequences involves MULTIPLE ROUNDS OF RANDOM MUTATION followed by MULTIPLE selection steps as outlined above.
In a beautiful discussion of the distinction between these two models, British biologist Richard Dawkins (The Blind Watchmaker New York, 1986) simulated the creationists' straw-man caricature on a computer. He programmed the computer to generate random sequences to see if it would ever generate a line from Hamlet: "Methinks it is a weasel." This line has 28 characters (including spaces), so the computer was programmed to make 28 selections from the 27 possible characters (26 letters plus space). A typical output was
MWR SWTNUXMLCDLEUBXTQHNZVJQF
Since there are 2728 different possible ways of choosing from 27 alternatives 28 times, one can calculate the probability of picking the correct sequence and, based on the speed of the computer, estimate how long on average one would have to wait for the correct sequence to be printed. Dawkins figured a million million million million million years. If this were the best way protein evolution could be conceptualized -- by selection in a SINGLE step from random sequences -- one might conclude, along with the creationists, that a protein sequence could not have evolved. But the creationists' single step selection model is clearly a "straw-man" designed to ridicule the concept of randomness as a component of evolution. The real evolutionist model is that modern amino acid sequences evolved by successive steps in which random mutations of pre-existing sequences were subjected to selection; any rare mutant that provided more efficient function was propagated to future generations, in which the process of mutation and selection was repeated over and over. When Dawkins terminated his computer program simulating the straw-man "creationist version" of evolution and rewrote a program that more closely approximates the "evolutionist version" of evolution, the results of the simulation were quite different. Dawkins programmed the computer to generate an initial sequence randomly, as in the first model, and the computer produced:
WSLMNLT DTJBKWIRZRESLMQCO P
Then, following Dawkins's revised program, the computer made multiple copies (progeny) of this sequence, while introducing random "errors" (mutations) into the copies. The computer examined all the mutated progeny and selected the one that had most similarity (however slight) to the line from Hamlet. This selected sequence was used as the basis for another generation of progeny with further mutations, from which the best copy was again selected -- and so on. By ten generations, the sequence had "evolved" to
MDLDMNLS ITJISWHRQREZ MECS P
By thirty generations, it was:
METHINGS IT ISWLIKE B WECSEL
Instead of taking millions of years, the computer generated METHINKS IT IS LIKE A WEASEL in about half an hour, at the forty-third generation. Thus a cumulative multi-step model is not at all implausible as a model for evolution, given both a mechanism for replicating imperfect copies and a strong selective pressure. (The replication mechanism is, of course, a big "given"; how such a mechanism might have developed is a separate question concerning the origin of life rather than its evolution, and is not the subject of this article.) The importance of Dawkins's simulation is that it highlights the error of all the creationist arguments against the statistical improbability of evolution, by showing that the creationists' choice of a single-step versus cumulative multi-step model creates a falsely low estimate of the potential for deriving a particular sequence via random mutation and selection. Although both the single-step model and the cumulative multi-step model involve random sequences and selection, the predicted consequences of the two models are very different. The creationists ignore this difference and intentionally discuss only the model that gives the result they like, even though this model corresponds least well to the theory of evolution.
Creationist Duane Gish has ridiculed Dawkins's computer model with the criticism that the final line from Hamlet was achieved only through an intelligently designed program running on an intelligently designed complex computer; the need for intelligent design to achieve a complex sequence is, says Gish, exactly what the creationists have been claiming from the beginning. (See also this web page.) At creation-vs-evolution debates, this argument is an effective creationist ploy since audiences generally can't see the fallacy quickly enough before the debate moves on to other topics. On reflection, however, it is clear that Gish's argument is false because it conflates the need for intelligent design in performing the simulation (a need that no one denies) with the need for design in what is being simulated; and it is the latter that is the issue at question. To make this distinction clear, consider the following example. Suppose a computer model of weather conditions is able to predict a rainstorm; no one will reasonably deny that the computer and the weather simulation software were intelligently designed, but this does not imply that the storm itself was created by a designer. The designed computer program is only an analytical tool for studying the natural process, and the necessity of intelligence for the analysis says nothing about the necessity of intelligence in the natural process. Similarly, the intelligent design that went into Dawkins's computer and simulation program do not at all imply that the processes being simulated -- random mutation and selection -- require intelligent design. (For a discussion of other criticisms of the Dawkins simulation, see the box above.)
Despite the logical fallacy in the creationists' dismissal of Dawkins's simulation, the seductive appeal of this argument led me to think that it could be most clearly countered if one could cite a biological example in which -- without the intervention of any intelligent designer -- successive rounds of mutation and selection could be unambiguously shown to lead to increased fitness within living organisms. As it happens, my own laboratory research in the area of antibody genes made me familiar with experiments showing just such a biological example. This example -- somatic mutation and selection of antibody genes -- should make it very difficult for the creationists to continue to insist that random mutations are always harmful and cannot lead to improved function in a real biological system. To appreciate the beauty of the mutational evolution of antibody genes, it is necessary to understand as background the deep mystery that this system posed before recombinant DNA technology made it possible to probe antibody genes directly, beginning in the late 1970s. (The discussion that follows may contain more information than you ever wanted to know about antibody genes, but this material is necessary to really understand the biology of the evolutionary model I will describe. For those readers who wish to skip over this background, the heart of the argument is in sections 4 and 5.)
It is common knowledge that a child who gets measles and then recovers is immune to further attacks by this virus. In fact, immunity to this or other illnesses can be generated in the absence of disease if various weakened forms of viruses, bacteria, and bacterial toxins are administered as vaccines. The protection that results from such vaccines is not due to a general strengthening of the body's defenses, but rather is highly specific; inoculation with a vaccine based on a particular strain of bacterium or virus protects against that agent, but often does not protect against infection by even closely related strains. Furthermore, experiments of the last century demonstrated that in many cases the immunity depends upon specific proteins that are present in blood after vaccination. These proteins, called antibodies (or immunoglobulins) can bind specifically to molecules from the foreign material (or antigen) in the vaccine. The binding of antibody to antigen can kill invading bacteria, neutralize invading viruses or toxins, and target all of these foreign materials for destruction by "garbage-eating" white blood cells. Antibodies are secreted into the bloodstream by a another kind of white blood cell called the B lymphocyte.
If one takes blood samples from an animal at different times before and after immunization by injecting an antigen, one generally finds that before immunization the blood does not contain significant amounts of antibody specific for the antigen. Beginning several days after immunization, antibody against the injected antigen begins to increase in the blood, often peaking at one to two weeks after immunization. A subsequent injection of the same antigen (a "booster") producers a much faster response, with higher amounts of antibody.
An important feature of the interaction between a particular antibody and its antigen is the tightness of the binding between these two molecules; this tightness, which can be measured by experiments, depends on how good the fit is between the antibodies and antigen, analogous to the fit of hand in glove or key in lock. In general, during the course of an immune response, the antibodies increase not only in numbers but also in the tightness with which they bind antigen -- their "affinity." The affinity often rises still further on subsequent booster shots of antigen. By binding more tightly to antigen, high affinity antibodies are much more efficient in carrying out their protective tasks.
Antibodies were found early on to be proteins -- that is they are made of amino acids whose sequence determines their properties, including their antigen-binding specificity. The information governing exactly which amino acids are used for each position in any protein sequence is stored in the gene for that protein. For each gene, the sequence information is encoded chemically in the sequence of subunits (known as nucleotides) in the long linear molecule of deoxyribonucleic acid (DNA). (A more detailed discussion of DNA structure and function may be found in section 2.1 of my Plagiarized Errors and Molecular Genetics article.)
The recognition that our immune systems are capable of producing -- precisely when needed -- highly specific antibodies against an immense number of bacterial and viral antigens led to three profound mysteries: (1) How does the body realize exactly which antibody genes need to be activated to fight a specific infection so that it can produce just the right antibodies? (2) How does our DNA store the immense amount of information necessary to encode specific antibodies against all the foreign invaders that we may encounter? This mystery is compounded by seemingly conflicting estimates that a mouse has no more than 100,000 genes but can make more than a million different antibodies, each of which would seem to require its own gene. (3) How can the progressive increase in antibody affinity during an immune response be explained?
An answer to the first question was suggested by MacFarlane Burnet in a hypothesis known as the "clonal selection theory." (See Figure 1.) According to this model, each of the millions of B lymphocytes circulating in a resting state in the blood of an animal has the potential to become an active antibody-secreting cell; but each B lymphocyte can make only one species of antibody, with a particular amino acid sequence and thus a particular antigen specificity. Before immunization, each resting B lymphocyte displays on its surface a membrane-bound form of the antibody that it will be able to secrete if the cell is activated. When an antigen -- for example, polio virus -- is injected into an animal, it circulates among the lymphocytes in the body. The vast majority of resting B lymphocytes express surface antibodies that cannot bind polio; these cells cannot be activated by the virus and therefore remain in a resting state and do not secrete antibody. But the virus will bind to the rare B lymphocytes displaying antibodies that can bind to polio. Binding of the virus to these cells triggers them into action: they proliferate, producing many daughter cells -- clones -- all capable of making antibodies that can bind to the virus. Then these activated progeny cells turn into miniature factories pouring out large quantities of anti-polio antibody. This mechanism explains how each antigen can trigger the production of just those antibodies capable of binding to it. The clonal selection theory was verified through a series of elegant experiments in the 1960's (Ada & Nossal Scientific American 257;62, 1987).
|
|
|
Figure 1. Clonal selection theory. Before exposure to antigen, millions of lymphocytes (three are shown in the top panel) circulate around the body in a resting state. Each cell displays on its surface many copies of a Y shaped antibody molecule (although only one molecule is drawn for each cell in the figure), but each cell makes a slightly different antibody. If any cell encounters an antigen (shown as floating black triangles in the figure) that its antibody can bind, that cell becomes activated (as shown for the middle cell in the top panel). The activated cell proliferates into a clone of many daughter cells, each expressing the same antibody on its surface (middle panel). The activated cells in this clone then mature into cells that can secrete antibody molecules into the circulation (bottom panel); all these antibody molecules will be able to bind the antigen that initially stimulated the original B cell. The model described here is a somewhat simplified version of the established theory. |
The second question -- how the myriad antigen specificities are encoded in the immunoglobulin gene DNA -- was solved by sequence analysis of homogeneous antibodies and their genes. These studies revealed that each antibody molecule is composed of four protein chains (see Figure 2): two identical large proteins (heavy chains) and two identical smaller light chains. The amino acid sequences of these chains were found to have an unusual property: the first hundred or so amino acids of each chain form a domain that is different for virtually every antibody that is sequenced ("variable" or V region), while the rest of the sequence is identical for every antibody chain of a particular class ("constant" or C region). (Among light and heavy chains there are about ten different classes of antibody chains, but the distinctions between these classes are irrelevant to this discussion.) Not surprisingly, the variable domains are involved in binding to the diverse possible antigens. The second question considered above can then be reformulated: how can the diversity of amino acid sequences of the variable regions of antibody proteins be encoded in the DNA, and how do the constant regions stay constant in the face of such variable region diversity?
|
|
|
Figure 2. Constructing antibodies and their genes. The top panel shows how the Y-shaped antibody molecule (at left) is composed of two identical light chains and two identical heavy chains; the individual protein chains are pictured in the middle and right of this panel. In this panel the portions of each protein that have constant (C) sequences are shown in black, while the variable (V) regions are gray or hatched. The middle panel shows the DNA structure of the antibody variable region genes in their assembled form found in B lymphocytes. The light chain variable gene is composed of two elements -- VL and JL -- joined together, whereas the heavy chain gene is composed of VH, D and JH elements. The bottom panel shows how the elements that form the light and heavy variable region genes are organized before they are assembled in B lymphocytes. A cluster of separate VL regions lies some distance in the DNA away from the cluster of J region genes. Similarly the VH, D and JH region genes form separate clusters. Each B lymphocyte joins one VL and one JL to form the assembled light chain variable region, and one VH, one D and one JH to form the assembled heavy chain variable region gene. |
These questions yielded a truly amazing answer, which is described in simplified outline as follows. It turns out that the gene that encodes each antibody variable region is created within each B lymphocyte by DNA rearrangements that join elements that are separated in all the non-lymphoid cells of the body (Tonegawa, Nature 302:575, 1983; see Figure 2, middle and bottom panels). Thus lymphocytes are an exception to the general rule that all cells of the body have the same DNA sequence. A heavy chain variable region gene is made by assembling three elements -- known as VH, D and JH -- and the comparable light chain variable region is made of two elements: VL and JL. These five types of elements are often referred to as "germline" elements, since they are separated in the DNA of germ cells (egg and sperm). Each human B lymphocyte can choose, Chinese menu style, one VH (out of about 50), one DH (out of 23), one JH (out of 6), one VL (out of 57) and one JL (out of 9); but there is only one gene for each class of the constant regions, so these domains do not vary for all antibodies of a given class. The number of possible different combinations of these germline elements is impressively large, but the number of possible antibodies is even larger because extra sequence variation occurs where the germline elements are joined together. The repertoire from the variable gene assembly process -- an estimated 30 million possible amino acid sequences from less than 200 separate genetic elements -- makes it likely that, for most foreign antigens, there will be antibody on the surface of some B lymphocytes that can bind the antigen, with low affinity perhaps, but enough to initiate an immune response. The elucidation of the unique DNA rearrangements that underlie antibody gene generation solved the second of the three mysteries of antibody formation, how millions of antibody structures can be generated in organisms having only about 100,000 genes. For his contribution to solving this problem, Susumu Tonegawa was awarded the 1987 Nobel Price in Physiology and Medicine..
It is in considering the third and last question -- how antibody affinity increases during an immune response -- that we come to the raison d'etre of this article, for investigations have clearly shown that the mechanism of the affinity rise that progressively improves the efficiency of antibody function is random mutation and selection. The evidence comes from analysis of several immune responses in inbred strains of mice, which all paint the same general picture (Wysocki et al. Proc Natl Acad Sci USA 83:1847, 1986; Griffiths et al. Nature 312:271, 1984). The responses are analyzed by determining the antibody gene structures from B lymphocytes taken before and at different times after immunization with an antigen (see Figure 3). Before immunization and during the first few days after immunization, any antigen-binding cells express antibody gene sequences derived from unaltered combinations of the germline elements described above. But beginning after about a week, the sequences clearly show evidence of mutation: many sequences are different from the germline elements from which they were constructed. Because the animals in these experiments are inbred, all the individuals are like identical twins, born with identical DNA sequences in all their genes. In particular, the sequences of their germline variable region gene elements are all known, so that any immunoglobulin sequences differing from the corresponding germline genes must be the result of mutations that occurred during the development of the B lymphocyte, i.e. somatic mutations.
|
|
|
Figure 3. The evidence for somatic mutation. In this type of experiment, an inbred mouse is injected with an antigen. After 1 week or 2 weeks (or more) B lymphocytes are obtained from the mice and the sequence of the relevant antibody gene (the light chain gene VL-JL is pictured in the figure) is determined and compared to the corresponding germline element sequences. After one week all the antibody gene sequences are identical to those of the germline elements, but by two weeks, multiple mutations (indicated by various shaped flags in the figure) can be observed. Mutations that are shared between several different genes (black symbols) allow construction of a genealogical tree (right) that depicts the presumed sequence of mutational events. In this genealogy, the progenitor B cell is shown at the top. Distinct mutations occur in various lines leading to progeny expressing some shared and some unique mutations. Frequently -- and mirroring the situation in phylogenetic evolution -- sequences representing "transitional" B cells at the precise divergence point between two sublineages (the cells marked A and B in the figure), are not recovered; but their existence and sequence can be deduced from the pattern of shared mutations among the available sequences. |
This somatic mutation process has several interesting properties. It occurs only in B lymphocytes and only when these cells are at a particular developmental stage and location in the "germinal center" of lymphoid tissues (Jacob et al., Nature 354:389, 1991). The process increases the normally low level of mutation (due to rare errors in the copying of DNA) by more than a thousand-fold, and has therefore been termed "hypermutation." The mutations occur almost exclusively in antibody genes, though recent data suggest that some other genes that are specifically expressed in germinal center B cells may also be affected (Shen et al, Science 280:1750, 1998). In antibody genes the mutations are found only in the region of an assembled variable domain gene, and not in the constant region (Lebecque and Gearhart J Exp Med 172:1717, 1990) or in unassembled variable region genes not expressed in a given B lymphocyte (Gorski et al., Science 220:1179, 1983). Yet, aside from their clustering near the assembled variable region genes, the mutations appear to be random. Different mutations occur in different cells, without a clear pattern in the nucleotide changes. Although some "hotspots" have been noted, i.e. short regions that show higher than average mutations (Levy et al. J Exp Med 168:475, 1988), most of the mutations are scattered around the targeted variable region gene. Some mutations do not alter the amino acid encoded in the gene; indeed, some fall completely outside the coding region of the gene in nearby "spacer" DNA where they can have no effect on the antibody produced by the cell. Scientists have been able to isolate and sequence mutated antibody genes from a single animal (Clarke et al., J Exp Med 161:687, 1985), or even from individual lymphocytes isolated from a single germinal center (Kuppers et al, EMBO J 12:4955, 1993). With this information one can construct a genealogical tree of antibody sequences -- much like the diagrams of species divergence that illustrate evolutionary genealogies -- by assuming that progeny of a particular B lymphocyte underwent several successive rounds of mutation (see Figure 3, right). Accordingly, identical mutations appearing in several independent sequences reflect mutational events that occurred early in the hypermutation process in an ancestral cell, while mutations unique to one sequence must have occurred in the later generations of that cell. The mutated antibodies isolated late in an immune response generally show higher binding affinity for antigen. In some cases the functional effects of individual mutations have been analyzed by engineering antibodies with different subsets of the mutations observed in a high affinity antibody. By comparing of the affinities of these engineered antibodies, scientists can deduce which mutations contributed to the increased affinity and which were incidental.
The model deduced from these findings provides an unambiguous biological example of the power of random mutations and selection. (The current model is slightly more complex than described below, but does not differ in any features relevant to the logic of this essay.) When antigen enters the body, it triggers a small number of B lymphocytes -- namely those whose surface antibody can bind the antigen -- to multiply and secrete antibody. These early responding antibody sequences are made of assembled germline gene elements in unmutated form, and frequently have low affinity. As the immune response continues and B cells move to germinal centers, hypermutation initiates, and begins to generate antibodies with altered structure. The hypermutation mechanism acts randomly and independently in the different clonal progeny cells, introducing random alterations in the antibody sequence in each cell. Most cells undergoing hypermutation end up producing antibody with unaltered or reduced affinity for the antigen; the latter cells would no longer be activated by antigen. However, rare mutations lead to antibodies of higher affinity for antigen. As the existing antibodies help to remove progressively more antigen from the circulation and the antigen concentration falls, selection for high affinity becomes the crucial factor in determining which cells will be stimulated by antigen. With lower amounts of antigen present, the cells expressing low affinity antibody on their surface become progressively less able to bind and be stimulated by antigen; in the environment of the germinal center, these poorly stimulated B cells are programmed to die by a specific process known as "apoptosis." (Choe et al, J Immunol 157:1006,1996) In contrast, the cells with high affinity antibody continue to bind antigen, and thus continue be stimulated to proliferate and secrete antibody. As the antigen concentration progressively falls while mutation and selection continue, the intensity of the selective pressure for high affinity increases. Repeated cycles of mutation and selection can lead to affinity levels 100-fold higher than that of the original unmutated antibody. The "competition" for efficient antigen binding has been shown to be the selective force driving the rise in antibody affinity, since if antigen is repeatedly administered to prevent the drop in antigen level and thereby eliminate the selective pressure for efficient antigen binding, antibody affinity does not rise (Eisen and Siskind, Biochemistry 3:996, 1964). Furthermore, when selection pressure has been experimentally removed by engineering mice with impaired capacity for programmed death by apoptosis, many B cells are found that make mutated antibodies with low affinity (Takahashi et al. J Exp Med. 190:39, 1999).
Late in the course of an immune response, as antigen becomes completely cleared from the bloodstream the amount of antibody secreted gradually falls and the immune response ends; but a subset of the last group of highly efficient cells persists as a quiescent population known as "memory cells," ready to respond with rapid secretion of high affinity antibody should they ever be triggered by another encounter with the same antigen in the future.
Clearly what we observe in the antibody response is evolution in miniature. In this model we can learn the structure of a gene at the beginning of the experiment and observe the accumulation of randomly induced mutations under natural selection for progressively improved function. This model of evolution is similar to the computer simulation discussed earlier, but it has two advantages as a persuasive example. First, it is a natural biological phenomenon rather than a theoretical designed simulation. And second, as in real phylogenetic evolution, the selection pressure is for biological function rather than for a specific target sequence chosen by an intelligent "creator." Thus the different sets of mutations observed in different high affinity antibodies that bind the same antigen represent alternative solutions to a particular selective challenge, just as different globin sequences in different species represent alternative solutions to the need for an oxygen-carrying protein.
Obviously there are differences between this kind of antibody evolution and the phylogenetic evolution that produced the diversity of plants and animals that we find on our planet. But none of these differences critically weaken the logic of the analogy between these two kinds of evolution as examples of random mutation and selection. Both involve sequences altered by random mutations, including rare beneficial alterations that "take over" the population because of their increased efficiency in proliferating under selective pressure; then these mutants are themselves "taken over" by later mutations, leading to progressively more efficient structures.
Thus the molecular immunogenetics evidence of antibody evolution that I have described makes it clear that, contrary to the creationists' claims, the combination of random mutation and selection CAN be a potent creative biological engine for the generation of progressive functional improvements. This evidence alone does not prove that life evolved as Darwin suggested, but it highlights the emptiness of another invalid, though superficially appealing, creationist objection to evolution: the false idea that random mutation is a uniformly deleterious process that could never be the source of improved biological function. And, to people who can appreciate the amazing complexity of life as a thing of wonder, the story of the generation of antibody diversity reveals in the immune system another example of an undesigned but beautifully functioning system.
The evolution of antibody gene affinity by mutation and natural selection has not been widely cited to counter the creationist claim that mutations can never lead to improved fitness. However, I have presented this argument at several debates with creationist spokesman Duane Gish of the Institute for Creation Research, who received a Ph.D. in biochemistry. Below I consider the two objections raised by Dr. Gish to the antibody evolution argument.
As described below, Gish has rejected the idea of somatic mutation of antibody genes, stating at a public debate with me that "a sick person would die" before high-affinity mutated antibodies could evolve. This claim reveals Gish's ignorance of immunology. There are many immune mechanisms other than antigen-specific antibodies that protect us in the early stages of infection before the highest affinity antibodies are made. These include cell-mediated immunity as well as the mechanisms referred to as "innate," "non-specific," or "non-adaptive" immunity, which include effects of C-reactive protein, maltose-binding protein, NRAMP-1, cytokine-induced macrophage activation, peptides such as magainins and defensins, etc. Moreover, "natural antibody," a mixture of antibodies present in normal serum in the absence of an intentional immunization, can confer some protection against certain infections before significant somatic mutation can occur. In fact, patients who make no antibodies at all as a result of a genetic defect are able to handle some types of infection without great difficulty. For all these reasons, Gish is completely wrong in his idea that the notion of somatic mutation predicts that we would die from trivial infections before high affinity antibodies evolve.
In private correspondence with me after our last debate, Gish indicated another reason why he disbelieved the mutation/selection model of antibody evolution: he felt that if the model were true then random mutations might lead to an antibody that could bind to the body's own molecules, causing an immune attack on our own tissues or "auto-immunity." Certainly auto-immune diseases exist; and in several autoimmune diseases that have been studied, mutated antibodies indeed appear to play a pathogenic role. However, somatic mutation does not routinely cause these diseases because the immune system has several complex mechanisms that prevent auto-immune responses. These protective mechanisms go under the general name of "tolerance," and include clonal deletion, receptor editing, anergy, veto cells, and suppressor cells, although we do not yet have a complete understanding of tolerance. Auto-immune diseases can occur when the tolerance mechanisms somehow fail and allow the production of anti-self antibodies, whether generated by somatic mutation or variable gene assembly recombination. But apparently in most individuals, the tolerance mechanisms are efficient enough to prevent mutated genes encoding auto-antibodies from causing pathology; cells harboring such genes are inactivated, forced to change their expressed antibody gene, or killed. Because of the effectiveness of tolerance mechanisms, the benefits of increased antibody affinity achieved by somatic mutation outweigh the risks of auto-immunity.
At my debates with Dr. Gish I have stressed my view that "creation science" is actually pseudoscience, and that the failure of its proponents to present their arguments in the peer-reviewed scientific literature reveals the status of their scholarship to be on par with that of dowsers, UFO enthusiasts and believers in a "Flat Earth." Whatever success creationists have had in promoting "creation science" depends on presenting their case before naive audiences untrained in science, who have insufficient background to recognize the false logic and false claims on which creationism rests. Gish always counters that creationists are excellent scientists whose work does not get chosen for publication in mainstream science journals only because of the prejudice of journal editors and reviewers against creationists. But at the debates I point out numerous examples of poor scholarship by creationists that completely explain why their efforts don't meet the standards of excellence for scientific publication. Gish's attempts to rebut the model of antibody evolution have presented a revealing example of his own scientific scholarship.
When confronted with the argument about evolution of antibody affinity in several of our debates, Gish simply denied that somatic mutation of antibody genes occurs. He claimed that the idea is controversial and that he was surprised that I believed it. In his own account of our most recent debate, Gish writes: "He [Gish] stated flatly that a sick person would die long before random chance mutations could ever produce the necessary antibodies to fight off an infection, and that the body has a mechanism for synthesizing antibodies precisely designed to protect it." Gish's account of the debate omits what happened next. I pointed out that the study of antibody genes was my field of scientific expertise, and that I was aware of many experiments described in the scientific literature that provided abundant evidence for the phenomenon, but I knew of no evidence against it. I challenged Gish to explain the flaws in these published experiments or to cite a single scientific study that contradicted them. Needless to say, Gish could offer no support for his claim that somatic mutation is controversial. I then lamented the fact that Dr. Gish could claim expertise in biochemistry and yet deny a phenomenon so important and well-accepted that it is taught to first-year biochemistry students; I had found discussions of antibody mutation in all five introductory biochemistry textbooks that I examined in a recent visit to a local bookstore. Gish responded that the question of antibody genes was a deep mystery and that anyone who solved it would get the Nobel prize. I pointed out that a Nobel Prize had in fact been awarded to Susumu Tonegawa several years ago for exactly that achievement. Gish appeared to be ignorant not only of somatic mutation but also of the basic biochemistry of antibody genes that received considerable publicity in newspapers, magazines and TV when Tonegawa's award was announced.
How would a legitimate scientist -- adhering to the normal standards of scientific scholarship and honesty -- have acted if confronted with an argument that he was unfamiliar with and that challenged his own views? He would, in my opinion, have admitted his ignorance of this topic and deferred judgment until he could examine the details of the science; and after the debate he would have immediately checked the literature relating to the new argument to see if his own views needed correcting. In contrast, after Dr. Gish heard from me about somatic mutation of antibody genes in one debate, and after denying that somatic mutation occurred, he apparently failed to investigate the published scientific literature on this issue, since he made the same false argument in subsequent debates. I feel that Dr. Gish's willingness to argue against an expert in a field Gish knows almost nothing about, shamelessly bluffing in front of a naive and trusting audience, makes clear the low standard of scientific scholarship of the most vocal spokesman of "creationist science." And given this low standard, there is no need to hypothesize prejudice to explain why his and other creationist arguments are rejected by the scientific community. Other examples documenting the low level of Gish's scientific scholarship are found at the following URLs:
I welcome Email responses from readers. This posting will be updated as necessary to accommodate such responses.
A draft of this essay was sent to Dr. Gish for comments; he wrote back that he would provide a rebuttal, but that he was too busy to respond immediately.

Home Page | Browse |
Search | Feedback | Links
The
FAQ | Must-Read Files | Index | Creationism | Evolution | Age of the
Earth | Flood Geology | Catastrophism | Debates
![[Fig1]](http://www.talkorigins.org/faqs/fitness/fig1.gif)
![[Fig2]](http://www.talkorigins.org/faqs/fitness/fig2.gif)
![[Fig3]](http://www.talkorigins.org/faqs/fitness/fig3.gif)